Прионите са протеини, които могат да съществуват в две форми – нормална и с променена структура (погрешно нагъната). Прионите с погрешно нагъната форма имат свойството да променят структурата на нормалните приони.
Няколко приона с променена структура могат да образуват популация, което има смъртоносни последици. Грешно нагънатият PrP прион води до фатални невродегенеративни заболявания при хора и животни. Прионите с променена структура са инфекциозни агенти – малко количество от тях може да зарази и унищожи цял организъм.
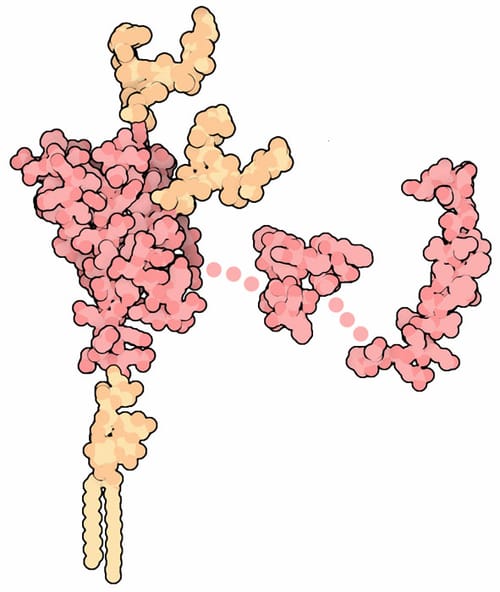
Приони – нормална и инфекциозна форма
Клетъчният прионен протеин (PrPC) присъства във всички ядрени клетки, въпреки че се произвежда главно в нервните клетки. При хората PrPC се кодира от PRNP гена, който е локализиран на късото рамо на хромозома 20.
PrPC има невропротективни функции, участва в невротрансмисията, в определянето на съдбата на стволовите клетки (в какъв вид зрели клетки ще се превърнат) и в редица други процеси. Прионът може да съществува в две триизмерни състояния: физиологична форма (нормална) и т.нар. изоформа, наречена скрейпи прионен протеин – PrPSc.
Тази форма на приона е инфекциозна и води до редица заболявания, като скрейпи при овце („луда овца“), хронично изтощение при елените, известно като заболяването „зомби елен“, спонгиформна енцефалопатия по говедата („луда крава“), а при човека – болест на Кройцфелд–Якоб (CJD), фатална фамилна инсомния и синдром на Герстман–Щрауслер–Шайнкер. Погрешното нагъване на PrPC в амилоидогенната изоформа PrPSc е ключово патогенно събитие при прионните заболявания, които могат да бъдат с генетичен характер, инфекциозни (чрез заразяване) или спорадични (при случайно образуване на патогенната форма).
Анормалните изоформи се натрупват в мозъка, което води до образуването на амилоидни плаки. Подобен механизъм за образуване на амилоидни плаки е добре установен при невродегенеративни заболявания, като болест на Алцхаймер, болест на Паркинсон, амиотрофична латерална склероза, фронтотемпорална деменция и болест на Хънтингтън.
Какво представляват прионните заболявания
Повечето човешки прионни заболявания са спорадични (85%), следвани по честота от генетичните (10–15%), и накрая са придобитите прионни заболявания вследствие на консумация на замърсено говеждо месо или от замърсени неврохирургични инструменти (~1%). Диагнозата на човешката прионна болест се постига чрез оценка на клиничната картина, изключване на други потенциални причини и използване на определени диагностични тестове, които могат да подскажат наличие на заболяването.
Изследването на цереброспинална течност също се използва за диагностициране на прионна болест. Ядрено-магнитният резонанс е полезен и има диагностична чувствителност 70–95% и специфичност приблизително 98% за спорадичен CJD в зависимост от приложените критерии.
Генетично прионно заболяване се установява чрез откриване на патогенен вариант в PRNP гена, а спонтанно възникналият CJD се диагностицира чрез разпознаване на известни рискови фактори, например трансплантация на роговица. Освен че играят ключова роля в генетичните прионни заболявания, вариациите в PRNP последователностите също са важни за спорадични и придобити прионни заболявания.
Пътят към лечението на прионните заболявания
Тези заболявания се срещат изключително рядко, но за тях няма лечение и винаги завършват с летален изход. Важно е да бъдат изследвани, за да се предотвратят епидемии, причинени от консумация на заразено месо от селскостопански животни.
Годишно има между един или два нови случая на 1 милион души, предимно при хора над 60-годишна възраст. Търсенето на лечение на прионните заболявания започва още през 1982 г., когато неврологът Стенли Прусинър от Калифорнийския университет в Сан Франциско идентифицира инфекциозните приони като причина за невродегенеративни заболявания.
В последното рандомизирано, контролирано от плацебо клинично изпитване, фокусирано върху прионните заболявания, се използва вече тествана стратегия при други невродегенеративни болести: фрагмент от синтетична ДНК, наречен антисенс олигонуклеотид (ASO), който може да достигне до мозъка чрез инжектиране в гръбначния мозък и да унищожи информационната РНК (иРНК), съществена за производството на протеин, причиняващ заболяването.
ASO в новото изпитване, произведен от Ionis Pharmaceuticals и наречен ION717, е насочен към иРНК, кодираща нормален прионен протеин, без който не може да възникне неправилно нагънатата форма. Две други компании имат за цел да премахнат нормалния прионен протеин чрез различни методи. Sangamo Therapeutics разработва т.нар. протеинови цинкови пръсти (ZFP), пригодени да се свързват с ДНК на пациента и да спират експресията на гена за приони. Те ще бъдат доставени като ДНК, кодираща цинкови пръсти, опаковани в безвреден вирус и приложени като еднократна интравенозна инжекция. Междувременно Gate Bioscience разработва перорално лекарство, което ще блокира производството на прионен протеин в мозъчните клетки.
{kind=link}
Какво да очакваме в бъдеще
Извършването на генетично изследване винаги трябва да се обсъжда със засегнатите от прионна болест семейства, независимо от наличието нa фамилна анамнеза. Има няколко възможности, препоръчителни за хора в риск, които желаят да имат биологични деца без PRNP мутации.
Извършването на генетичните тестове може да се увеличи, ако пациентите вземат участие в предстоящи клинични изпитвания. Генетичните подходи към лечението може да се използват за предотвратяването или забавянето на клиничното начало при носители на мутации и за лечението на пациенти със симптоматично заболяване. Генните терапии имат за цел да намалят PrPc с цел премахване на субстрата за превръщане на прионите в патогенни форми. Необходима е още работа, за да се оцени как генетичните терапии могат да повлияят на човешките прионни заболявания.
Помогнете ни да научим какви са читателските ви възприятия и отношението ви към „Тоест“, като попълните нашата анкета.
„Тоест“ се издържа единствено от читателски дарения
Ако харесвате нашата работа и искате да продължим, включете се с месечно дарение.
Подкрепете ни